
W skrócie:
|
Na czym polega zespół Angelmana?
Zespół Angelmana to uwarunkowane genetycznie współwystępowanie u jednej osoby chorób neurologicznych, w tym:
- specyficznych zaburzeń zachowania,
- upośledzenia funkcji poznawczych i komunikacji werbalnej,
- nieprawidłowego rozwoju w sferze ruchowej,
- oraz specyficznych cech w zakresie fenotypu morfologicznego.
Pierwszego opisu pacjentów pediatrycznych z charakterystycznym wyglądem twarzy dokonał w 1965 roku brytyjski pediatra Harry Angelman.
Trudno jest jednoznacznie oszacować częstość występowania Zespołu Angelmana. W zależności od źródeł raportowano 1 na 10 000/12 000 urodzeń (2015). Chorobowość mieści się w przedziale 1:10 000 – 1:62 000.
Czynniki ryzyka wystąpienia zespołu Angelmana
Do wystąpienia charakterystycznych objawów Zespołu Angelmana prowadzą różne zaburzenia molekularne regionu q11–q13 chromosomu 15. W związku z powyższym ocena ryzyka ujawnienia się choroby jest skomplikowana. Ponad 98 proc. przypadków Zespołu Angelmana wynika z losowych zdarzeń genetycznych i nie jest dziedziczona od rodzica.
Prawdopodobieństwo ponownego pojawienia się chorego dziecka w takich rodzinach nie przekracza 1 proc. Tylko u części przypadków Zespołu Angelmana, będących następstwem obecności mutacji w genie UBE3A u matki, istnieje 50 proc. ryzyko urodzenia chorego dziecka w odniesieniu do każdej ciąży.
| Należy podkreślić, że przekazanie zmutowanego genu UBE3A przez matkę, powoduje manifestację cech związanych z zespołem Angelmana, natomiast przekazanie uszkodzonego genu UBE3A przez ojca, nie wywołuje u dziecka objawów choroby. |
Zespół Angelmana objawy
Po porodzie stan ogólny dziecka z Zespołem Angelmana oceniany jest jako dobry. Do ukończenia 3. miesiąca życia rozwój dziecka zazwyczaj przebiega prawidłowo. Nie obserwuje się takich odchyleń jak:
- niezadowalający przyrost masy ciała,
- przedłużająca się żółtaczka,
- nieprawidłowe napięcie mięśni,
- ubogie ruchy kończyn,
- drżenie kończyn,
- zaburzenia oddychania,
- czy brak odruchów fizjologicznych.
Pomiędzy 6. miesiącem a 2. rokiem życia zauważalne jest opóźnienie rozwoju psychoruchowego u dziecka z Zespołem Angelmana. Zauważalne stają się pierwsze objawy choroby, takie jak:
- trudności w karmieniu,
- obniżone napięcie mięśniowe,
- upośledzone nabywanie umiejętności ruchowych zwłaszcza w zakresie dużej motoryki,
- dzieci zaczynają siadać około 2. roku życia, a chodzić około 5. roku życia.
W 1995 roku opracowano kryteria diagnostyczne zespołu Angelmana (aktualizacja 2005 rok). | ||
Grupa I | Grupa II | Grupa III |
Cechy obecne u wszystkich osób z Zespołem Angelmana:
| Cechy, które występują często u około 80% osób z Zespołem Angelmana:
| Cechy współtowarzyszące:
|
* Dzieci z zespołem Angelmana są pogodnie usposobione, przez co często określane są jako „dzieci marionetki” z uwagi na charakterystyczny uśmiech, zaburzone ruchy kończyn i napady śmiechu.
Zespół Angelmana przyczyny
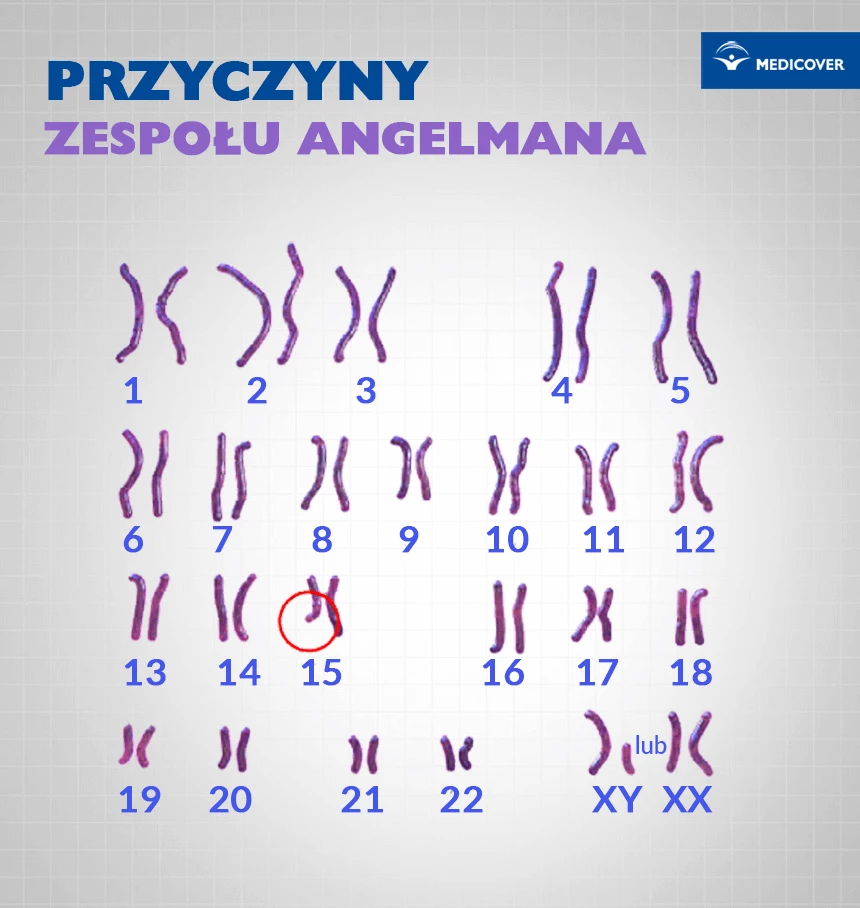
Zespół Angelmana wywołany jest utratą funkcji genu UBE3A pochodzącego od matki, który zlokalizowany jest na jednym z chromosomów 15 pary. Do takiej sytuacji może dojść na 4 różne sposoby (mechanizmy):
I - delecja w regionie q11–q13 matczynego chromosomu 15 (70–75% przypadków),
II - ojcowska trisomia chromosomu 15, czyli zdarzenie molekularne, gdy oba chromosomy z pary homologicznej pochodzą od ojca (2–5%),
III – defekt imprintingu matczynego w tym rejonie (3-5%),
IV – mutacja w genie UBE3A (10%).
Podłoże molekularne Zespołu Angelmana jest złożone i wynika z właściwości genu UBE3A, który ulega imprintingowi, czyli piętnowaniu genomowemu w komórkach nerwowych mózgu.
Piętnowanie genomowe zwane inaczej naznaczeniem rodzicielskim polega na tym, że pewne geny pochodzące od ojca i matki wykazują odmienną aktywność (ekspresję) w zarodku i wszystkich komórkach potomnych. Najczęściej regulacja ekspresji genów odbywa się na drodze metylacji DNA.
Gen UBE3A znajdujący się na chromosomie 15 odziedziczonym od ojca jest nieaktywny w mózgu, natomiast gen odziedziczony od matki ulega ekspresji w neuronach (odbywa się na nim synteza ważnego dla neuronów enzymu E6-AP ligazy).
Jak diagnozowany jest zespół Angelmana?
Zespół Angelmana diagnozowany jest na podstawie:
- obrazu klinicznego pacjenta,
- analizy rodowodu,
- oraz badań molekularnych i dodatkowych.
Weryfikacja rozpoznania klinicznego Zespołu Angelmana opiera się na odpowiednio przeprowadzonej analizie DNA:
I. Detekcja mikrodelecji (ubytku materiału genetycznego) techniką FISH.
II. Badanie techniką acgh (metoda porównawczej hybrydyzacji genomowej do mikromacierzy). Technika aCGH umożliwia ocenę całego genomu z wysoką rozdzielczością.
III. Analiza stopnia metylacji regionu 15q11.2-q13.
IV. Sekwencjonowanie całego genu UBE3A.
V. Ocena markerów DNA w celu ujawnienia ojcowskiej disomii jednorodzicielskiej.

Zespół Angelmana rokowania
Zespół Angelmana należy do całościowych i przewlekłych zaburzeń rozwoju, wymagających wielospecjalistycznej terapii i opieki przez całe życie pacjenta. Obecnie nie dysponujemy leczeniem przyczynowym. Naturalny przebieg Zespołu Angelmana jest ściśle powiązany z jego podłożem molekularnym.
Badania korelacji pomiędzy fenotypem a rodzajem defektu DNA wskazują, że delecje fragmentu chromosomu 15 odpowiadają za cięższą postać kliniczną, niż mutacje w genie UBE3A, co objawia się w postaci:
- znacznej niepełnosprawności intelektualnej,
- wyraźnych zaburzeń mowy,
- ujawnienia się padaczki w młodszym wieku,
- częstszych występowań mikrocefalii,
- bardziej nasilonej dysmorfii w obrębie twarzy,
- występowania zaburzeń psychicznych,
- czy zahamowania wzrostu.
Jak długo żyje osoba z zespołem Angelmana?
Obecnie szacuje się, że długość życia osób z Zespołem Angelmana jest podobna jak w populacji ogólnej. Istnieją udokumentowane przypadki pacjentów, którzy osiągnęli 70. rok życia.
Czy są testy prenatalne na zespół Angelmana?
Wszystkie choroby, które można rozpoznać postnatalnie (po porodzie), można również identyfikować w okresie prenatalnym.
Przeczytaj także: |
Dowiedz się więcej
Dieta z niskim indeksem glikemicznym w kontroli napadów padaczkowych w zespole Angelmana
http://angelman.org.pl/wp-content/uploads/2022/04/Low_Glycemic_Index_Treatment_AS.pdf
System opieki nad dzieckiem z zespołem Angelmana w Stanach Zjednoczonych - doświadczenia matek
https://repozytorium.bg.ug.edu.pl/info/article/UOG36cc69cf1e914a1c9a0b05b94b01868d/
Źródła:
Williams C. et al.: Angelman syndrome: mimicking conditions and phenotypes. Am J Med Genet 2001; 101:59–64. 12.
Minassian B.A. et al.: Angelman syndrome: correlations between epilepsy phenotypes and genotypes. Ann Neurol 1998; 43:485–93.
Williams C.A. et al.: Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am. J. Med. Genet. 1995; 56: 237–238.
Petersen M.B. et al.: Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. Am. J. Med. Genet. 1995; 60: 261–262.
Kantor B. et al.: Control elements within the PWS/AS imprinting box and their function in the imprinting process. Hum Mol Genet. 2004; 13:751–762.
Goodin K. et al.: Postępy w badaniach genetycznych i ich zastosowanie w diagnostyce klinicznej noworodków, 2009, Vol. 13 Nr 4 Pediatria po Dyplomie, 67-78.

Prezentowanych informacji o charakterze medycznym nie należy traktować jako wytycznych postępowania medycznego w stosunku do każdego pacjenta. O postępowaniu medycznym, w tym o zakresie i częstotliwości badań diagnostycznych i/lub procedur terapeutycznych decyduje lekarz indywidualnie, zgodnie ze wskazaniami medycznymi, które ustala po zapoznaniu się ze stanem pacjenta. Lekarz podejmuje decyzję w porozumieniu z pacjentem. W przypadku chęci realizacji badań nieobjętych wskazaniami lekarskimi, pacjent ma możliwość ich odpłatnego wykonania. Należy potwierdzić przy zakupie badania szczegóły do jego przygotowania. |